Publié le
Lecture 14 mins
Les tumeurs cutanées malignes de l’enfant
Sylvie FRAITAG, Anatomo-cytopathologiste, hôpital Necker-Enfants malades, Paris
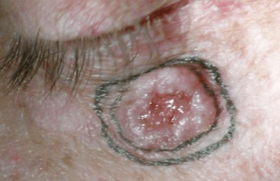
Les tumeurs malignes de l’enfant sont rares mais représentent une problématique majeure pour le clinicien. Le diagnostic précoce permettant la mise en route d’un traitement adapté est l’un des facteurs essentiels du pronostic. Ainsi, la réalisation d’une biopsie s’impose au moindre doute, en l’absence d’un diagnostic clinique certain.
Les tumeurs cutanées malignes sont rares chez l’enfant et ne représentent que 1,4 % des tumeurs ; les plus fréquentes étant représentées par les hamartomes et les tumeurs bénignes. Environ deux tiers des tumeurs malignes sont primitives, et un tiers sont métastatiques avec, par ordre de...
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :